> Адреногенитальный синдром


- Details
- Hits: 748
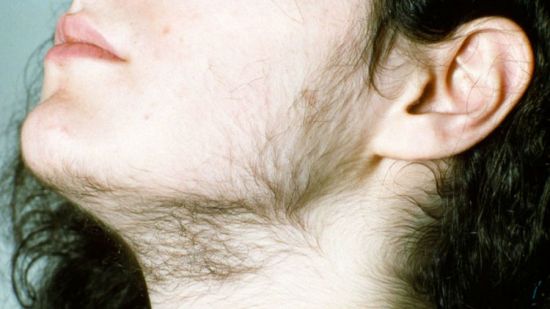
Адреногенитальный синдром врожденное заболевание, характеризующееся прогрессирующей вирилизацией, ускоренным соматическим развитием, повышенной экскрецией гормонов коры надпочечников. Наиболее часто встречается тип гиперплазии надпочечников, вызванный дефицитом 21-гидроксилазы. Дефицит 21-гидроксилазы приводит к нарушению образования дезоксикортикостерона и 11-дезоксикортизола. Биосинтез андрогенов не нарушен, что приводит к их избыточной продукции еще при внутриутробном развитии. У новорожденных девочек отмечается разная степень маскулинизации от умеренной гипертрофии клитора до полного срастания губно-мошоночных складок с формированием предстательной железы, мошонки, полового члена с отверстием мочеиспускательного канала. Внутренние половые органы сформированы правильно, кариотип 46,ХХ. Отмечается гиперпигментация околососковой и генитальной областей. У мальчиков основными клиническими симптомами являются раннее половое развитие и низкий рост, связанный с преждевременным закрытием зон роста эпифизов.
Для формы адреногенитального синдрома, составляющей 1/3 случаев, характерны данные признаки, электролитный баланс, уровень кортизола и альдостерона в норме. Сольтеряющая форма составляет 2/3 классических случаев адреногенитального синдрома. Кроме вирилизующего действия избытка андрогенов наблюдается нарушение солевого обмена в результате дефицита минералокортикоидов и вовлечения в патологический процесс ренин-альдостероновой системы. Заболевание проявляется в первые дни жизни срыгиванием, рвотой, потерей веса, сонливостью, признаками дегидротации, гипонатриемией, гиперкалиемией, метаболическим ацидозом. При поздней (неклассической) форме адреногенитального синдрома отсутствуют симптомы вирилизации у девочек при рождении. Первые признаки появляются в подростковом возрасте. Для девочек характерно умеренное увеличение клитора, ускорение костного возраста, нарушение менструальной функции, маскулинное телосложение. У мальчиков признаки менее заметны, единственными симптомами могут быть ускорение костного возраста и преждевременное половое оволосение. Рост взрослых ниже среднего, фертильность снижена. Во всех случаях заболевания отмечается повышение в моче уровня 17-кетостероидов и прегнантриола. Тип наследования аутосомно-рецессивный. Популяционная частота 1:1 000 000 неклассическая форма, 1:18 000 классическая форма. Ген, мутации в котором приводят к развитию АГС картирован на хромосоме 6р21.3. Ген 21-гидроксилазы CYP21OHB, CYP21А2) и псевдоген 21-гидроксилазы (CYP21OHA) организованы в тандем повторов с генами С4В и С4А комплемента между генами HLA-B и HLA-DR. Из-за высокой степени гомологии между повторами неравные рекомбинации и генная конверсия ответственны за большинство мутаций в этом локусе. Ген CYP21A2 состоит из 10 экзонов, имеет длину 6.3 т.п.н. Ген и псевдоген гомологичны на 98%. Гипотетический продукт псевдогена полностью не функционален. 30% обнаруженных мутаций составляет делеция гена CYP21A2. Эта мутация ответственна за развитие сольтеряющей формы заболевания. Другой мутацией, ответственной за развитие сольтеряющей формы, является мутация сайта сплайсинга во втором интроне (35%). Большинство из других мутаций в норме присутствует в псевдогене CYP21OHA и, вероятно, представляют собой результат генной конверсии.
К сольтеряющей форме заболевания приводят мутации Q318X, кластер миссенс-мутаций в экзоне 6 (I235N/V236E/ M238K). Для вирильной формы характерна мутация I172N. За развитие неклассической формы заболевания с поздним началом ответственны, в большинстве случаев, мутации P30L, V281L, P453S. В Западной Европе 60% мутаций при неклассической форме АГС составляет мутация V281L. Данный анализ проводится для больных детей с обязательным предоставленим материала их родителей, материал родителей может понадобиться для исследования т.к. при конверсии больших участков псевдогена последовательность гена CYP21А2 может включать в себя несколько мутаций. Поэтому при проведении анализа необходимо определить, находятся ли мутации на одной хромосоме или лежат на разных хромосомах. Наличие материала родителей поможет в этих случаях определить расположение мутаций на хромосомах.
> Синдром Ретта
- Details
- Hits: 1716
Синдром Ретта X-сцепленное доминантное заболевание, вызванное делецией локуса Xq28 или мутациями гена МЕСР2, лежащего в данном локусе. Ген МЕСР2 состоит из 4 экзонов и кодирует метил-СрG-связывающий белок 2. Мутации в этом гене отвечают за 90-95% случаев развития классического синдрома Ретта и 40-60% случаев атипичных форм синдрома Ретта. Белок MeCP2 принадлежит к семейству метил-связывающих протеинов. Он связывается со специфическими генами-мишенями быстро развивающегося мозга и регулирует их эксперссию, влияя, таким образом, на развитие нервной ткани. В гене описано около 800 мутаций, большинство из которых являются нуклеотидными заменами и локализованы в экзонах 3 и 4. Девять точковых мутаций являются наиболее частыми (R168X, R270X, R255X, T158M, R306C, R294X, R133C, R106W, L386fs), доля которых составляет 60-70%, около 10% мутаций это небольшие делеции (20-150 н.п.) в карбокси-терминальном сегменте гена МЕСР2, крупные перестроения, захватывающие целые экзоны или даже ген составляют от 7,5% в атипичных случаях до 37% при классической форме.
Частота заболевания 1:10000-1:15000. Случаи синдрома Ретта, как правило, носят спорадический характер, представляя собой мутации de novo, однако описаны и семейные случаи (при гонадном мозаицизме у одного из родителей). Данное заболевание является второй по частоте после синдрома Дауна причиной тяжёлой умственной отсталости у девочек. Для плодов гемизиготных по Х-хромосоме мутации в гене МЕСР2 являются летальными. Поэтому у мужчин синдром Ретта практически не встречается, за исключением случаев синдрома Кляйнфельтера и соматического мозаицизма.

При классической форме синдрома Ретта раннее психомоторное развитие ребенка протекает нормально, но в возрасте 6-18 месяцев развитие приостанавливается и в дальнейшем происходит регресс приобретенных двигательных навыков, речи, социальных контактов, появляются сереотипные движения рук (потирание, моющие движения и т.п.), судороги, происходит задержка умственного развития. У больных наблюдается нарушение походки, мышечная гипотония, микроцефалия, сколиоз. После регресса происходит некоторое улучшение или стабилизация состояния.
Помимо классического синдрома Ретта, существуют атипичные формы: вариант Запела (variant Zapella) с сохраненной речью, форма с поздним началом регресса, Х-сцепленная синдромальная умственная отсталость у мальчиков (тип Луба), тяжелая врожденная энцефалопатия у мальчиков и др. Мутации в гене МЕСР2 обнаруживают при некоторых синдромах, характеризующихся задержкой умственного развития, таких как Ангельман-подобный синдром, синдром, схожий с синдромом ломкой Х-хромосомы, фенотипы схожие с аутизмом. За развитие некоторых атипичных форм, таких как, ранняя эпилептическая энцефалопатия 2, врожденная форма синдрома Ретта, отвечают другие гены (CDKL5, FOXG1).
В Центре AFGEN Молекулярной Генетики методом количественной MLPA проводится поиск делеции / дупликации локуса Xq28 и прямое автоматическое секвенирование гена МЕСР2.
> Болезнь Вильсона
- Details
- Hits: 683
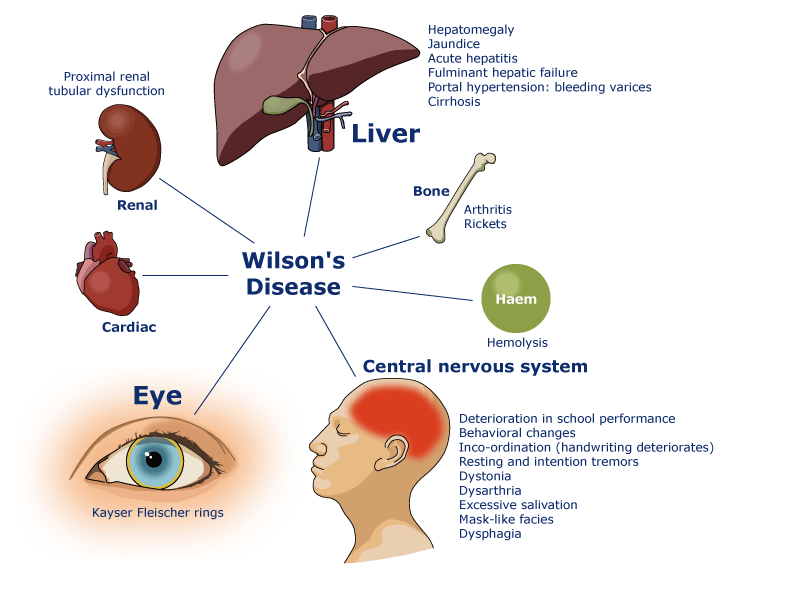
Болезнь Вильсона — Коновалова (Гепатоцеребральная дистрофия, гепатолентикулярная дегенерация, болезнь Вестфаля — Вильсона) — врождённое нарушение метаболизма меди, приводящее к тяжелейшим наследственным болезням центральной нервной системы и внутренних органов. Диагностируется у 5-10 % больных циррозом печени дошкольного и школьного возраста. Заболевание передаётся по аутосомно-рецессивному типу. Ген ATP7B, мутации которого вызывают заболевание, расположен на 13-й хромосоме (участок 13q14-q21).
Аутосомно-рецессивный тип наследования болезни Вильсона. 25 % вероятность рождения больного у родителей-гетерозигот
Ген болезни Вильсона — Коновалова(ATP7B) расположен в длинном плече 13-й хромосомы. Ген кодирует P-тип АТФазы, которая транспортирует медь в жёлчь и включает её в церулоплазмин. В 10 % случаев мутация не обнаруживается.
Хотя описано почти 300 мутаций ATP7B, в большинстве популяций болезнь Вильсона возникает в результате небольшого количества мутаций, специфичных для этих популяций. Например, для западных популяций мутация H1069Q (замена гистидина на глутамин в позиции 1069 белка) присутствует в 37-63 % случаев заболевания, в то время как в Китае эта мутация очень редка и R778L (замена аргинина на лейцин в позиции 778) встречается чаще. Относительно мало известно о влиянии мутаций на течение заболевания, хотя по данным некоторых исследований мутация H1069Q предполагает более позднее начало неврологических симптомов.
Нормальные вариации в гене PRNP могут изменить течение болезни, увеличивая возраст появления заболевания и влияя на тип симптомов, которые развиваются. Этот ген кодирует прионный белок, который активен в головном мозге и других тканях, а также, как полагают, участвует в транспорте меди.
У заболевания аутосомно-рецессивный тип наследования. То есть больной должен получить дефектный ген от обоих родителей. Люди только с одним мутантным геном называются носителями (гетерозиготы). У них могут возникать слабовыраженные нарушения метаболизма меди.
> Атаксия Фридрейха
- Details
- Hits: 992
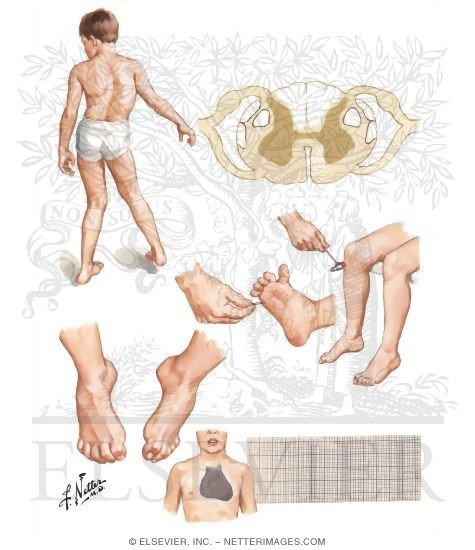
Атаксия Фридрейха (FRDA) является наследственной нейродегенеративной патологией, классически характеризующейся прогрессирующей атаксией походки и конечностей, дизартрией, дисфагией, глазодвигательными нарушениями, потерей глубоких сухожильных рефлексов, пирамидными знаками, сколиозом, и в некоторых случаях, кардиомиопатией, сахарным диабетом, потерей зрения и нарушениями слуха. Распространенность FRDA у представителей белой европеоидной расы оценивается от 1/20,000 до 1/50,000. Классические проявления FRDA начинаются в детстве или юности. Общая неуклюжесть и атаксия походки обычно - первые признаки, часто сопровождаемые пирамидальными признаками, атаксией верхних конечностей и дизартрией. Окуломоторные признаки появляются рано и включают в себя нестабильность фиксации (square wave jerks) и нистагм. Потеря зрения может произойти позже. Слуховая нейропатия (8-39% случаев), приводит к нарушению слуха. Интеллектуальное развитие кажется незатронутым. Арефлексия и дистальная потеря чувствительности присутствуют в большинстве случаев. Дисфагия сначала проявляется в легкой форме, но при прогрессировании болезни может привести к поперхиванию твердой и жидкой пищей. Сколиоз и деформации ног (полая стопа и эквиноварусная деформация стопы) могут быть легкой или тяжелой степени. Мышечная спастичность, отмечающаяся позже в течении заболевания, может вызывать дискомфорт, причинять боль, проблемы позиционирования и контрактуры у некоторых больных. Поражение сердца (типичная гипертрофическая кардиомиопатия), обычно развивается после неврологических проявлений и главным образом бессимптомно. Сахарный диабет, замеченный максимум в 30% случаев, часто проявляется позже. Сообщается о гиперактивности мочевого пузыря в некоторых случаях. Среднее время от начала симптомов до необходимости инвалидного кресла составляет 15.5 лет (диапазон 3 - 44). Было описано несколько нетипичных фенотипов, но совпадение признаков существенное.
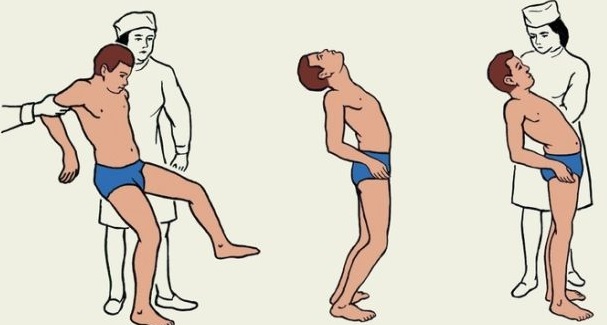
Первые симптомы обычно появляются на первом, втором десятилетии жизни, однако первые проявления могут быть и на третьем и четвертом десятилетии. Проявляется атаксия при ходьбе, а также нарушением почерка, дизартрией, слабостью в ногах, нарушением или потерей слуха. Нарушается глубокая чувствительность, постепенно нарастает мышечная атрофия, на начальных этапах более выраженная на нижних конечностях, но с течением болезни захватывающая и верхние. Атрофируется зрительный нерв, развивается катаракта, что ведёт к слепоте, нарушается функция тазовых органов, развивается деменция.
Развиваются нарушения со стороны эндокринной системы — сахарный диабет, гипогонадизм, дисфункция яичников, кардиомиопатии. Возникают скелетные деформации — искривление позвоночника, «стопа Фридрейха».
Лечение
Лечение симптоматическое; уделяют внимание развивающемуся сахарному диабету, нарушениям со стороны сердечно-сосудистой системы. Хирургическая коррекция возникающих деформаций опорно-двигательного аппарата.
> Расстройства аутистического спектра
- Details
- Hits: 807
Расстройства аутистического спектра проявляются в диапазоне состояний, которые характеризуются определенным нарушением социального поведения, коммуникации и вербальных способностей и сужением интересов и деятельности, которые одновременно специфичны для индивидуума и часто повторяются. Расстройства аутистического спектра начинаются в детстве и сохраняются в подростковом и взрослом возрасте. В большинстве случаев эти состояния проявляются в первые 5 лет жизни.
РАС часто сопровождаются другими нарушениями, в том числе эпилепсией, депрессией, тревожным состоянием и гиперактивным расстройством с дефицитом внимания. Интеллектуальный уровень крайне варьируется: от тяжелого повреждения до высоких когнитивных способностей.

Лица с расстройством аутистического спектра испытывают проблемы в социальной коммуникации: например, не могут поддержать диалог, испытывают проблемы социального сближения, не могут делиться интересами, эмоциями, всё это может доходить до неспособности начинать или реагировать на социальные взаимодействия[1]:50.
Поддержание и понимание социальных взаимоотношений также страдает: от трудности с приобретением друзей, трудностей с участием в играх, в которых задействовано воображение, в крайних случаях доходя до видимого отсутствия интереса к любым контактам со сверстниками.
Также присутствуют проблемы в невербальном коммуникативном поведении: аномалия зрительного контакта (больным трудно поддерживать зрительный контакт, более того, он может вызывать неприятные ощущения), языка тела или телесной ориентации, речевой интонации, или проблемы с использованием и пониманием сути невербального общения. Лица с РАС могут выучить несколько функциональных жестов, но их репертуар меньше, чем у других людей, спонтанно в общении они не могут адекватно использовать жесты. В крайних случаях доходит до полного отсутствия зрительного контакта, мимики или жестов.
Характерны крайне ограниченные и зацикленные на одном интересы (также может присутствовать сильная привязанность к необычным предметам).
Патологическая реакция на входную сенсорную информацию (например, видимое безразличие к температуре окружающей среды, безразличие к боли, негативная реакция на определённые звуки или шумы).
Стереотипность, повторяемость поведения, интересов или деятельности в целом. Это может проявляться в стереотипных движениях, у детей — в выстраивание игрушек в строгом порядке, идиосинкразических фразах или эхолалии (стереотипном повторении фраз других людей). Чрезмерная потребность в неизменности и постоянности (например, неизменный распорядок дня, неизменный маршрут прогулки или однообразная еда)[1]:50. Многие взрослые с расстройством аутистического спектра учатся подавлять стереотипное повторяющееся поведение на публике, удаётся это у лиц без умственной отсталости и речевых затруднений. При присутствии коморбидного кататонического синдрома (кататонии) у лица с расстройством аутистического спектра, в DSM-5 используется дополнительный код 293.89/F06.1.
Для постановки диагноза по DSM-5 «расстройство аутистического спектра» перечисленные в этом разделе симптомы должны вызывать клинически значимое ухудшение в социальной, профессиональной или других важных сферах повседневного функционирования, иначе диагноз не ставится.

Симптомы РАС делят на две большие группы: социальную коммуникацию и повторяющиеся или ограниченные действия, занятия и интересы. Первая группа — это отсутствие интереса к общению, способности говорить или понимать других. Обычно именно эти симптомы помогают родителям заподозрить, что с ребенком что-то не так.
Ко второй группе симптомов относятся:
Повторяющиеся движения тела, например, хлопки, скручивания, качания, наклоны;
Последовательность действий и ритуалы, нарушение которых расстраивает человека;
Ограниченность интересов;
Слишком чуткое или наоборот притупленное восприятие запахов, звуков и прикосновений.
Обычно на симптомы обращают внимание в детстве — в возрасте от 2 до 3 лет. Но не всегда специалисты могут диагностировать РАС у детей, потому что симптомы могут проявляться у здоровых детей, которые испытывают стресс, тревогу или депрессию. Поэтому заболевание иногда обнаруживают только у взрослых.
Bizimlə əlaqə
- Ünvan: Bakı, 3 mkr.Pişəvari 110 (Cavadxan küç 24),Memar Əcəmi metrosunun yaxınlığında
- Tel: (+99412) / (+99450) / (+9955) / (+9970 )430 89 89
- (+9950) 265 66 99/265 66 00
- Email: info@afgen.az
Fotoqalereya






